Energy Decomposition to Access the Stability Changes Induced by CO Adsorption on Transition-Metal 13-Atom Clusters
Abstract
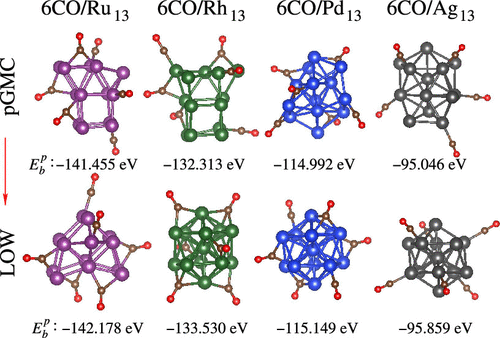
Our atomistic understanding of the physical–chemical parameters that drives the changes in the relative stability of clusters induced by adsorbed molecules is far from satisfactory. In this work, we employed density functional theory calculations to address this problem using CO adsorption on 13-atom transition-metal clusters, TM13, namely, nCO/TM13, where TM = Ru, Rh, Pd, and Ag, and n = 1–6. Unexpectedly, changes in the relative stability take place for all systems at a lower coverage, namely, at n = 3 (Ru13), 4 (Rh13, Ag13), and 2 (Pd13). To address the effects that lead to changes in the stability, we proposed an energy decomposition scheme for the binding energy of the nCO/TM13 systems, which yields that the change in relative stability is dominated by the interaction energy and cluster distortion energy upon adsorption, where the interaction energy is higher for high-energy unprotected clusters. Furthermore, we characterized all adsorption parameters, which helps us to complement our atomistic understanding.
Marcos G. Quiles
Associate Professor
My research interests include neural networks, machine learning, complex networks, and their applications in interdisciplinary problems, such as materials science and social networks.
Juarez L. F. Da Silva
Associate Professor
He works in the area of Computational Science of Materials and Chemistry using ab initio methods based on Density Functional Theory for the study of metallic surfaces, molecular adsorption, oxides, semiconductors, clusters, nanoparticles, and the development of algorithms for the global optimization of the nanoparticle structure.